New Resource: Comprehensive Review of 2024 Afirma Research Now Available
Download
Summary: New findings published in Cell offer a provocative hypothesis about the links between genetic variation and disease risk. While scientists have come to understand that large numbers of genes impact disease development, the prevailing theory has been that these genes are directly relevant to the disease in question. Now, a team of researchers at Stanford posits that not only are huge numbers of genes involved in complex traits and diseases like autism, schizophrenia and cancer, but that most of these genes are spread far and wide throughout the genome. They refer to this as an “omnigenic” model and, according to Veracyte scientists, its tenets may have important implications for thyroid cancer diagnosis.
Methodology: The Stanford team looked at height, a trait that shares a similar genetic architecture to diseases such as diabetes and atuimmune diseases and for which very large GWAS datasets are available. The authors also looked at active chromatin (e.g., RNA expression) data in several broadly defined cell-type groups, including immune, central nervous system and cardiovascular, from studies focused on Crohn’s disease, rheumatoid arthritis and schizophrenia. Looking further at the above three diseases, the authors also considered the contributions of genes from different functional areas.
Findings:
Distribution of GWAS Signals Across the Genome
Looking at data from multiple studies, they estimate that more than 100,000 single nucleotide polymorphisms (SNPs) impact height and that, while GWAS signals tend to be more enriched in predicted gene pathways, these causal variants are also spread widely across the genome and involve genes and pathways outside of those directly associated with height.
Enrichment of Genetic Signals in Transcriptionally Active Regions
The authors found that genetic contribution to disease is heavily concentrated in regions marked by active chromatin in relevant, but also in seemingly unrelated, tissue. They found no genetic contribution from inactive regions in these cell-type groups.
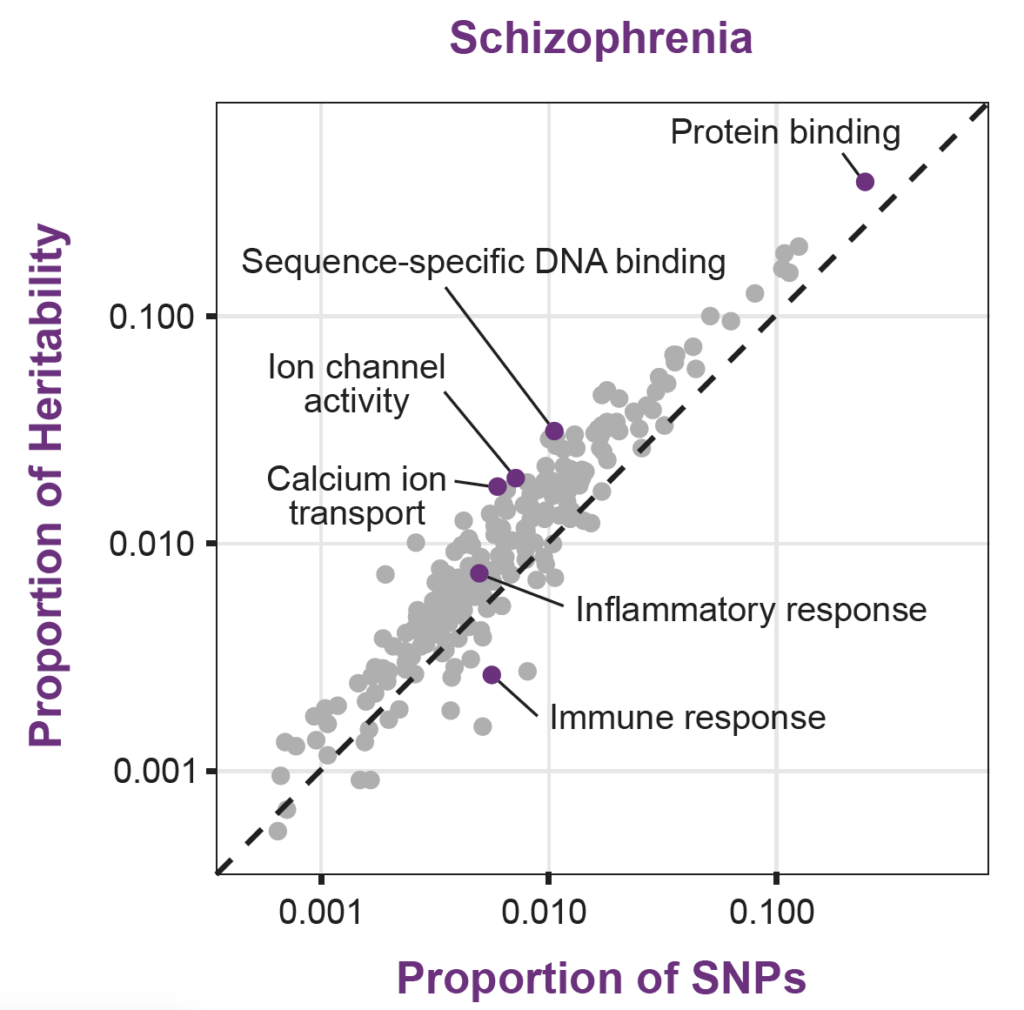
Figure 1. Gene Ontology Enrichments for Schizophrenia. Note that the diagonal indicates the genome-wide average across all SNPs. Analysis by stratified LD score regression (Finucane et al., 2015).
Weak Enrichment of Genetic Signals by Functional Categories
While genetic signals were more concentrated in functional areas relevant to the disease (e.g., inflammatory response and rheumatoid arthritis), they also found that genes in broad functional categories (e.g., protein binding) contributed more to total trait heritability.
Conclusions: The authors propose an “omnigenic” model in which most diseases and traits are directly affected by a modest number of genes or gene pathways – called “core genes” – that have specific roles in disease etiology. The researchers propose that cell regulatory networks are highly interconnected and that any gene expressed in disease-relevant cells is likely to affect the regulation or function of core genes. Further, these peripheral genes hugely outnumber core genes, meaning that a large fraction of the total genetic contribution to disease comes from peripheral genes that have no direct role in disease activity. The authors assert that, if their model is correct, then the ability to map these enormous networks of genes will be essential to fully understanding disease biology.
Commentary
by Giulia Kennedy, Ph. D.
Chief Scientific Officer and SVP of Research and Development at Veracyte
The “omnigenic” model proposed in this article suggests that most common diseases are associated with small effects from a
large number of genes, therefore implying that diseases such as thyroid cancer are unlikely to be accounted for by variants in a small number of genes. The model also shows that most of the contributions are derived from transcriptionally active portions of the genome, consistent with our work showing that the best classifiers rely heavily on gene expression to diagnose thyroid nodules. As the number of genes explaining thyroid cancer expand to large numbers, it becomes critical to use machine learning and statistical models to incorporate the small signals from many genes to derive an accurate diagnosis.